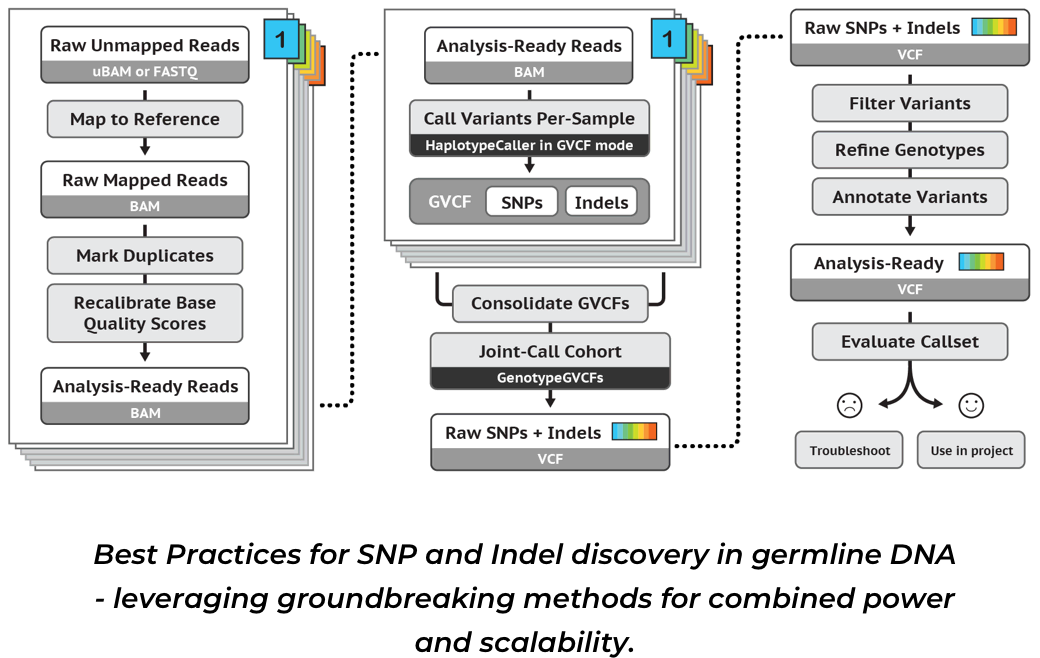
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
| #!/bin/bash
source activate GATK4
TASKN=SRX247249
MERGED=/home/jovyan/upload/merged/$TASKN
RefSeq=/home/jovyan/data/refseq/GRCh38.p14.fna
VCFPATH=$MERGED'/SAMPLE1'
knownSites=/home/jovyan/upload/knownSites
gatk VariantRecalibrator \
-R $RefSeq \
-V $VCFPATH'/HC.vcf.gz' \
--resource:hapmap,known=false,training=true,truth=true,prior=15.0 $knownSites/hapmap_3.3.hg38.ncbi.vcf.gz \
--resource:dbsnp,known=true,training=false,truth=false,prior=2.0 $knownSites/GRCh38.dbSNP.ncbi.vcf.gz \
-an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR \
-mode SNP \
-O $VCFPATH/snp.recal \
--tranches-file $VCFPATH/snp.tranches \
--rscript-file $VCFPATH/snp.plots.R
gatk ApplyVQSR \
-R $RefSeq \
-V $VCFPATH'/HC.vcf.gz' \
-O $VCFPATH'/snp.VQSR.vcf.gz' \
--truth-sensitivity-filter-level 99.0 \
--tranches-file $VCFPATH/snp.tranches \
--recal-file $VCFPATH/snp.recal \
-mode SNP
gatk VariantRecalibrator \
-R $RefSeq \
-V $VCFPATH'/snp.VQSR.vcf.gz' \
--resource:dbindel,known=true,training=false,truth=false,prior=2.0 $knownSites/Homo_sapiens_assembly38.known_indels.ncbi.vcf.gz \
--resource:mills,known=true,training=true,truth=true,prior=12.0 $knownSites/Mills_and_1000G_gold_standard.indels.hg38.ncbi.vcf.gz \
-an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR \
-mode INDEL --max-gaussians 6 \
-O $VCFPATH/snp.indel.recal \
--tranches-file $VCFPATH/snp.indel.tranches \
--rscript-file $VCFPATH/snp.indel.plots.R
gatk ApplyVQSR \
-R $RefSeq \
-V $VCFPATH'/snp.VQSR.vcf.gz' \
-O $VCFPATH'/snp.indel.VQSR.vcf.gz' \
--truth-sensitivity-filter-level 99.0 \
--tranches-file $VCFPATH/snp.indel.tranches \
--recal-file $VCFPATH/snp.indel.recal \
-mode INDEL
|