
2022-07-02-【迁移】STAR:一键脚本.md 14 KB
title: 【迁移】STAR:一键脚本 urlname: STAR--yi-jian-jiao-ben date: 2022-07-02 18:17:22
tags: STAR
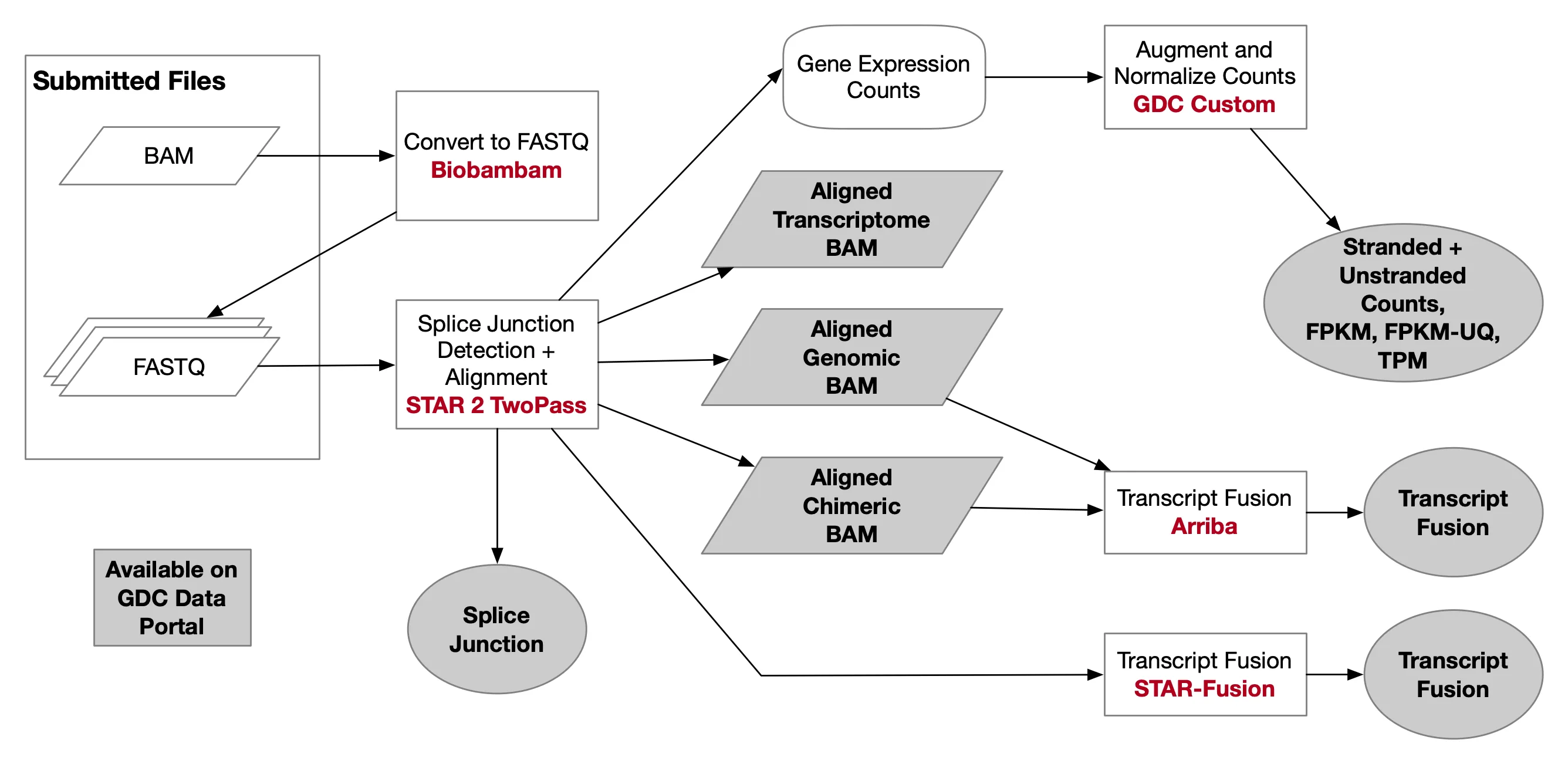
{% note info %} 2023-09-27更新:添加了转录本计数相关内容 {% endnote %}
一些依赖
安装 biobambam
项目地址:https://gitlab.com/german.tischler/biobambam2
conda create -n biobambam -c bioconda biobambam -y安装 star
conda create -n star -c bioconda star -y conda activate star conda install -c bioconda samtools -y conda install -c bioconda rsem -y
可选:BAM转FASTQ
将所有bam文件以.bam结尾,单独存放到一个目录,conda activate biobambam,新建以下脚本运行
#!/bin/sh
ROOT=/home/jovyan/upload/22.07.02/muscle
mkdir $ROOT
for _ in *.bam
do
mkdir $ROOT/${_%.bam}
bamtofastq \
collate=1 \
exclude=QCFAIL,SECONDARY,SUPPLEMENTARY \
filename=$_ \
gz=1 \
inputformat=bam \
level=5 \
outputdir=$ROOT/${_%.bam} \
outputperreadgroup=1 \
outputperreadgroupsuffixF=_1.fq.gz \
outputperreadgroupsuffixF2=_2.fq.gz \
outputperreadgroupsuffixO=_o1.fq.gz \
outputperreadgroupsuffixO2=_o2.fq.gz \
outputperreadgroupsuffixS=_s.fq.gz \
tryoq=1 \
done
- biobambam 高版本可能有 libmaus2 相关错误尚未修复
- filename、outputdir 等参数等于号后不能有空格
- 单端测序,outputdir 里只有 default_s.fq.gz 的输出
- 更多信息
首次:构建小鼠的基因组索引
来源
步骤
zcat XL789vs123/XL1/default_s.fq.gz | head #这个例子只有50, 因此 sjdbOverhang 为49
wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M29/GRCm39.primary_assembly.genome.fa.gz
wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M29/gencode.vM29.primary_assembly.annotation.gtf.gz
gunzip *.gz
conda activate star
nano 2.sh
chmod +x 2.sh
#!/bin/sh
STAR \
--runMode genomeGenerate \
--genomeDir index \
--genomeFastaFiles GRCm39.primary_assembly.genome.fa \
--sjdbOverhang 49 \
--sjdbGTFfile gencode.vM29.primary_assembly.annotation.gtf \
--runThreadN 8
- 如果要自己构建,可以使用 zcat R1.fq.gz | head 来查看reads长度,选用reads长度减1(149)作为 --sjdbOverhang 比默认的100要好,但是说明里认为绝大多数情况下100和理想值差不多
已构建好的索引
- TCGA 上有已经构建好的索引,但是可能是损坏的
wget https://api.gdc.cancer.gov/data/07f2dca9-cd39-4cbf-90d2-c7a1b8df5139 -O star-2.7.5c_GRCh38.d1.vd1_gencode.v36.tgz- tar -zxvf star-2.7.5c_GRCh38.d1.vd1_gencode.v36.tgz
附加 构建转录本索引
cd /home/jovyan/upload/zl_liu/star/ # gtf文件和genome.fa文件所在目录
rsem-prepare-reference --gtf gencode.v36.primary_assembly.annotation.gtf GRCh38.primary_assembly.genome.fa GRCh38.primary_assembly.rsem.idx -p 8
grep -e '^ENST' GRCh38.primary_assembly.rsem.idx.ti > GRCh38.primary_assembly.rsem.t2n.ti
grep -e '^ENSG' GRCh38.primary_assembly.rsem.idx.ti > GRCh38.primary_assembly.rsem.g2n.ti
大概生成了以下文件
├── [5.8M] ./GRCh38.primary_assembly.rsem.g2n.ti ├── [ 770] ./GRCh38.primary_assembly.rsem.idx.chrlist ├── [404K] ./GRCh38.primary_assembly.rsem.idx.grp ├── [362M] ./GRCh38.primary_assembly.rsem.idx.idx.fa ├── [362M] ./GRCh38.primary_assembly.rsem.idx.n2g.idx.fa ├── [386M] ./GRCh38.primary_assembly.rsem.idx.seq ├── [128M] ./GRCh38.primary_assembly.rsem.idx.ti ├── [362M] ./GRCh38.primary_assembly.rsem.idx.transcripts.fa ├── [6.5M] ./GRCh38.primary_assembly.rsem.t2n.ti一键脚本
单独起一个目录,在这个目录下,以样本名为目录名,将样本的fataq文件以gzip压缩后存放,单端一个文件,双端两个文件,都可以,示例结构如下
XL1_12/
├── XL01
│ └── default_s.fq.gz
├── XL02
│ └── default_s.fq.gz
├── XL03
│ └── default_s.fq.gz
├── XL04
│ └── default_s.fq.gz
├── XL05
│ └── default_s.fq.gz
├── XL06
│ └── default_s.fq.gz
├── XL07
│ └── default_s.fq.gz
├── XL08
│ └── default_s.fq.gz
├── XL09
│ └── default_s.fq.gz
├── XL10
│ └── default_s.fq.gz
├── XL11
│ └── default_s.fq.gz
└── XL12
└── default_s.fq.gz
在这个目录外,conda activate star,新建以下脚本运行,即可跑完前五步
#!/bin/bash
source activate star
#任务名
TASKN=XL1_12
#设置CleanData存放目录
CLEAN=/home/jovyan/upload/22.07.02/$TASKN
#设置输出目录
WORK=/home/jovyan/upload/22.07.02/output_$TASKN
#设置index目录
INDEX=/home/jovyan/upload/22.07.02/index
#设置参考文件位置
Reference=/home/jovyan/upload/22.07.02/GRCm39.primary_assembly.genome.fa
#设置 sjdbOverhang
sjdbOverhang=49
#设置 IIG 目录(这一步的输出目录)
IIG=/home/jovyan/upload/22.07.02/IIG_$TASKN
echo $CLEAN", "$WORK", "$INDEX
mkdir $WORK
CDIR=$(basename `pwd`)
echo $CDIR
echo $CLEAN
for file in $CLEAN/*
do
echo $file
SAMPLE=${file##*/}
echo $SAMPLE
mkdir $WORK"/"$SAMPLE
cd $WORK"/"$SAMPLE
STAR \
--genomeDir $INDEX \
--readFilesIn `ls $CLEAN/$SAMPLE/*` \
--runThreadN 4 \
--outFilterMultimapScoreRange 1 \
--outFilterMultimapNmax 20 \
--outFilterMismatchNmax 10 \
--alignIntronMax 500000 \
--alignMatesGapMax 1000000 \
--sjdbScore 2 \
--alignSJDBoverhangMin 1 \
--genomeLoad LoadAndRemove \
--readFilesCommand zcat \
--outFilterMatchNminOverLread 0.33 \
--outFilterScoreMinOverLread 0.33 \
--sjdbOverhang $sjdbOverhang \
--outSAMstrandField intronMotif \
--outSAMtype None \
--outSAMmode None \
done
echo $CLEAN", "$WORK", "$INDEX", "$IIG
mkdir $IIG
CDIR=$(basename `pwd`)
echo $CDIR
echo $CLEAN
for file in $CLEAN/*
do
echo $file
SAMPLE=${file##*/}
echo $WORK"/"$SAMPLE
done
STAR \
--runMode genomeGenerate \
--genomeDir $IIG \
--genomeFastaFiles $Reference \
--sjdbOverhang $sjdbOverhang \
--runThreadN 4 \
--sjdbFileChrStartEnd `ls $WORK/*/SJ.out.tab`
ln -s $INDEX/exonGeTrInfo.tab $IIG
ln -s $INDEX/exonInfo.tab $IIG
ln -s $INDEX/geneInfo.tab $IIG
ln -s $INDEX/sjdbList.fromGTF.out.tab $IIG
ln -s $INDEX/transcriptInfo.tab $IIG
CDIR=$(basename `pwd`)
echo $CDIR
echo $CLEAN
for file in $CLEAN/*
do
echo $file
SAMPLE=${file##*/}
echo $WORK"/"$SAMPLE
mkdir $WORK"/"$SAMPLE"/Res"
cd $WORK"/"$SAMPLE"/Res"
STAR \
--genomeDir $IIG \
--readFilesIn `ls $CLEAN/$SAMPLE/*` \
--runThreadN 8 \
--quantMode TranscriptomeSAM GeneCounts \
--outFilterMultimapScoreRange 1 \
--outFilterMultimapNmax 20 \
--outFilterMismatchNmax 10 \
--alignIntronMax 500000 \
--alignMatesGapMax 1000000 \
--sjdbScore 2 \
--alignSJDBoverhangMin 1 \
--genomeLoad LoadAndRemove \
--limitBAMsortRAM 35000000000 \
--readFilesCommand zcat \
--outFilterMatchNminOverLread 0.33 \
--outFilterScoreMinOverLread 0.33 \
--sjdbOverhang $sjdbOverhang \
--outSAMstrandField intronMotif \
--outSAMattributes NH HI NM MD AS XS \
--outSAMunmapped Within \
--outSAMtype BAM SortedByCoordinate \
--outSAMheaderHD @HD VN:1.4 \
--outSAMattrRGline ID:sample SM:sample PL:ILLUMINA
done
附加 转录本计数
- 转录本水平: cufflinks、eXpress、RSEM
外显子水平: DEXseq
nano RSEM.sh && chmod +x RSEM.sh ./RSEM.sh#!/bin/bash source activate star #任务名 TASKN=cleandata #设置上一步的输出目录 WORK=/home/jovyan/work/shRNA-seq/output_$TASKN #设置参考文件位置 Reference=/home/jovyan/upload/zl_liu/star/GRCh38.primary_assembly.genome.fa #设置转录本索引位置(前缀) RSEM_IDX=/home/jovyan/upload/zl_liu/star/GRCh38.primary_assembly.rsem.idx #设置这一步的输出目录 RSEM_OUT=/home/jovyan/work/shRNA-seq/RSEM_$TASKN mkdir -p $RSEM_OUT for file in $WORK/* do echo $file SAMPLE=${file##*/} echo $SAMPLE mkdir $RSEM_OUT"/"$SAMPLE cd $RSEM_OUT"/"$SAMPLE rsem-calculate-expression -no-bam-output --alignments -p 8 --paired-end \ $WORK"/"$SAMPLE"/Res/Aligned.toTranscriptome.out.bam" \ $RSEM_IDX \ 'resm' done最后得到的RSEM_OUT目录结构如下
[158M] . ├── [ 26M] ./NC-1 │ ├── [6.8M] ./NC-1/resm.genes.results │ ├── [ 15M] ./NC-1/resm.isoforms.results │ └── [4.3M] ./NC-1/resm.stat │ ├── [ 871] ./NC-1/resm.stat/resm.cnt │ ├── [493K] ./NC-1/resm.stat/resm.model │ └── [3.9M] ./NC-1/resm.stat/resm.theta ├── [ 26M] ./NC-2 │ ├── [6.8M] ./NC-2/resm.genes.results │ ├── [ 15M] ./NC-2/resm.isoforms.results │ └── [4.4M] ./NC-2/resm.stat │ ├── [ 872] ./NC-2/resm.stat/resm.cnt │ ├── [493K] ./NC-2/resm.stat/resm.model │ └── [3.9M] ./NC-2/resm.stat/resm.theta ├── [ 26M] ./NC-3 │ ├── [6.8M] ./NC-3/resm.genes.results │ ├── [ 15M] ./NC-3/resm.isoforms.results │ └── [4.4M] ./NC-3/resm.stat │ ├── [ 872] ./NC-3/resm.stat/resm.cnt │ ├── [493K] ./NC-3/resm.stat/resm.model │ └── [3.9M] ./NC-3/resm.stat/resm.theta ├── [ 26M] ./shRNA1-1 │ ├── [6.8M] ./shRNA1-1/resm.genes.results │ ├── [ 15M] ./shRNA1-1/resm.isoforms.results │ └── [4.4M] ./shRNA1-1/resm.stat │ ├── [ 874] ./shRNA1-1/resm.stat/resm.cnt │ ├── [493K] ./shRNA1-1/resm.stat/resm.model │ └── [3.9M] ./shRNA1-1/resm.stat/resm.theta ├── [ 26M] ./shRNA1-2 │ ├── [6.8M] ./shRNA1-2/resm.genes.results │ ├── [ 15M] ./shRNA1-2/resm.isoforms.results │ └── [4.3M] ./shRNA1-2/resm.stat │ ├── [ 849] ./shRNA1-2/resm.stat/resm.cnt │ ├── [493K] ./shRNA1-2/resm.stat/resm.model │ └── [3.8M] ./shRNA1-2/resm.stat/resm.theta └── [ 26M] ./shRNA1-3 ├── [6.8M] ./shRNA1-3/resm.genes.results ├── [ 15M] ./shRNA1-3/resm.isoforms.results └── [4.3M] ./shRNA1-3/resm.stat ├── [ 867] ./shRNA1-3/resm.stat/resm.cnt ├── [493K] ./shRNA1-3/resm.stat/resm.model └── [3.8M] ./shRNA1-3/resm.stat/resm.theta组装RSEM_Counts文件
#设置转录本索引转换的位置(前缀)(无idx) RSEM_x2n='/home/jovyan/upload/zl_liu/star/GRCh38.primary_assembly.rsem' #设置RSEM的输出目录 RSEM_OUT='/home/jovyan/work/shRNA-seq/RSEM_cleandata' file_list <- list.dirs(RSEM_OUT, recursive=F, full.names = F) t2n <- read.table(file = paste0(RSEM_x2n, '.t2n.ti') , sep = '\t', header = F) g2n <- read.table(file = paste0(RSEM_x2n, '.g2n.ti') , sep = '\t', header = F) transcript_counts <- cbind(t2n, g2n) colnames(transcript_counts) <- c('transcript_id', 'transcript_name', 'gene_id', 'symbol') transcript_tpm <- transcript_counts transcript_IsoPct <- transcript_counts for(sample in file_list){ test_tab <- read.table(file = file.path(RSEM_OUT, sample, 'resm.isoforms.results') , sep = '\t', header = T) transcript_counts[sample] <- test_tab$expected_count transcript_tpm[sample] <- test_tab$TPM transcript_IsoPct[sample] <- test_tab$IsoPct } write.csv(x = transcript_counts, file = 'transcript_counts.csv') # 期望计数 write.csv(x = transcript_tpm, file = 'transcript_tpm.csv') write.csv(x = transcript_IsoPct, file = 'transcript_IsoPct.csv') # 该转录本占所属基因的比例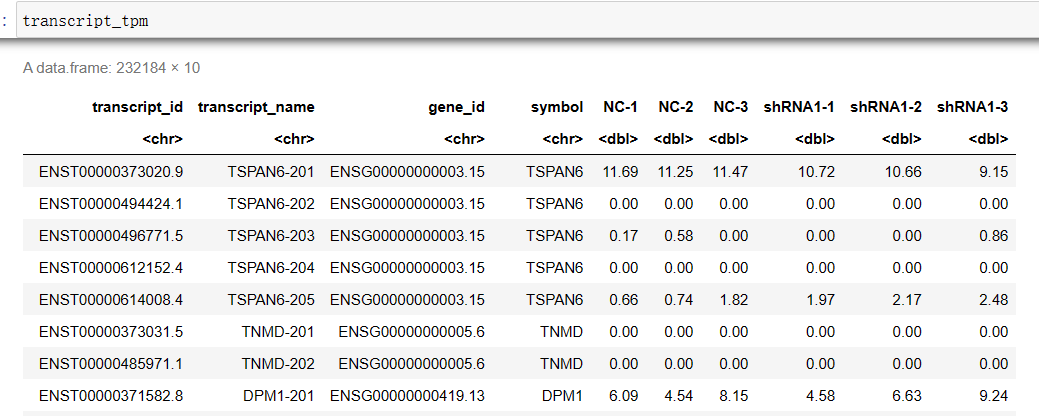
组装Counts文件
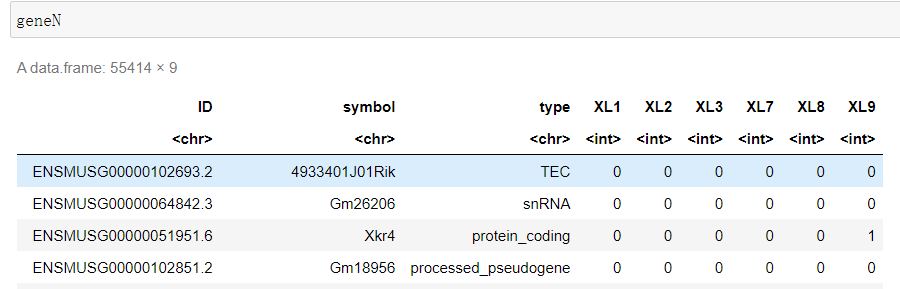
#设置第三步的输出目录 WORK='/home/jovyan/upload/22.07.02/output_XL789vs123' #设置index中基因注释位置 Reference='/home/jovyan/upload/22.07.02/index/geneInfo.tab' file_list <- list.dirs(WORK, recursive=F, full.names = F) geneN <- read.table(file = Reference, sep = '\t', skip = 1) colnames(geneN) <- c('ID', 'symbol', 'type') for(sample in file_list){ test_tab <- read.table(file = file.path(WORK, sample, 'Res', 'ReadsPerGene.out.tab') , sep = '\t', header = F) test_tab <- test_tab[-c(1:4), ] geneN[sample] <- test_tab[2] } write.csv(x = geneN, file = 'XL789vs123.csv')
DESeq2分析
library(DESeq2)
count_all <- read.csv("liver.csv",header=TRUE,row.names=1)
count_all
cts_b <- count_all[ ,c(-1,-2,-3)]
rownames(cts_b) <- count_all$ID
cts_bb <- cts_b
cts_b <- cts_bb[,c('XL07', 'XL08', 'XL09', 'XL10', 'XL11', 'XL12')]
keep <- rowSums(cts_b) > 10
cts_b[keep,]
conditions <- factor(c(rep("Control",3), rep("XL",3)))
colData_b <- data.frame(row.names = colnames(cts_b), conditions)
colData_b
dds <- DESeqDataSetFromMatrix(countData = cts_b[keep,],
colData = colData_b,
design = ~ conditions)
dds <- DESeq(dds)
res <- results(dds)
rres <- cbind(count_all[keep,c(1,2,3)], data.frame(res))
write.csv(rres, file='XL101112vs789_DESeq2.csv')
rres
附加:删除临时文件
#!/bin/sh
#设置CleanData存放目录
CLEAN=/home/jovyan/upload/yy_zhang(备份)/RNA-seq/Cleandata
#设置这一步的输出目录 (确保目录存在)
WORK=/home/jovyan/upload/zl_liu/star_data/yyz_01/output
for file in $CLEAN/*
do
echo $file
SAMPLE=${file##*/}
echo $SAMPLE
rm -rf $WORK"/"$SAMPLE"/IIG"
done