2023-09-21-【复习】单细胞最佳实践的流程.md 17 KB
title: 【复习】单细胞最佳实践的流程 urlname: dan-xi-bao-zui-jia-shi-jian-de-liu-cheng date: 2023-09-21 19:22:33
tags: ['单细胞', 'pipeline', 'DoubletFinder', 'DecontX']
配置环境
- 基础编程环境
-
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/") ## 指定 BiocManager::install 镜像 options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")) ## 指定 install.packages 镜像, options(ggrepel.max.overlaps = Inf)nano ~/.Rprofile # 内容如上,配置镜像 conda create -n seurat5 -c conda-forge r-seurat=4.3.0 -y conda activate seurat5 conda install -c conda-forge r-irkernel -y conda install -c conda-forge r-devtools -y conda install -c conda-forge r-biocmanager -y conda install -c conda-forge r-tidyverse -y conda install -c conda-forge r-clustree -y Rscript -e "IRkernel::installspec(name='seurat5', displayname='seurat5')" ## 开始 seurat5 的安装 wget https://github.com/satijalab/seurat/archive/refs/heads/seurat5.zip -O seurat5.zip Rscript -e "devtools::install_local('seurat5.zip')" ## Rscript -e "devtools::install_local('seurat5.zip', host = 'xxx.limour.top/token/api.github.com')" ## options(Seurat.object.assay.version = 'v5') 使用 seurat5 conda install -c conda-forge hdf5 -y wget https://github.com/bnprks/BPCells/archive/refs/heads/main.zip -O BPCells.zip Rscript -e "devtools::install_local('BPCells.zip')" ## 开始 DoubletFinder 的安装 wget https://github.com/chris-mcginnis-ucsf/DoubletFinder/archive/refs/heads/master.zip -O DoubletFinder.zip Rscript -e "devtools::install_local('DoubletFinder.zip')" ## 开始 decontX 的安装 wget https://github.com/campbio/decontX/archive/refs/heads/main.zip -O decontX.zip conda install -c conda-forge imagemagick -y conda install -c conda-forge r-ggrastr -y Rscript -e 'BiocManager::install("celda")' Rscript -e "devtools::install_local('decontX.zip')" ## api.github.com 错误就换用下面的命令 # Rscript -e 'remotes::install_local("decontX.zip", repos = NULL, type = "source")' # Rscript -e "devtools::install_local('decontX.zip', repos = NULL, type='source')" # Rscript -e 'install.packages("decontX.zip", repos = NULL, type = "source")' ## 开始 plot1cell 的安装 wget https://github.com/mojaveazure/loomR/archive/refs/heads/master.zip -O loomR.zip Rscript -e "devtools::install_local('loomR.zip')" wget https://github.com/HaojiaWu/plot1cell/archive/refs/heads/master.zip -O plot1cell.zip Rscript -e 'BiocManager::install(c("biomaRt","EnsDb.Hsapiens.v86","GEOquery","simplifyEnrichment"))' Rscript -e "devtools::install_local('plot1cell.zip')" ## 开始 SCTransform 的安装 Rscript -e 'BiocManager::install("glmGamPoi")'
附加 安装SCP
conda create -n r-scp -c conda-forge r -y
conda activate r-scp
conda install -c conda-forge r-irkernel -y
conda install -c conda-forge r-devtools -y
conda install -c conda-forge r-biocmanager -y
Rscript -e "IRkernel::installspec(name='r-scp', displayname='r-scp')"
## 开始 SCP 的安装
wget https://github.com/zhanghao-njmu/SCP/archive/refs/heads/main.zip -O SCP.zip
conda install -c conda-forge r-ggplot2 -y
conda install -c conda-forge r-gridgraphics -y
Rscript -e 'BiocManager::install("ggtree")'
Rscript -e "devtools::install_local('SCP.zip')"
## 找到 conda 的路径
whereis conda
# /opt/conda/bin/conda
## 创建 SCP 的 python 环境
options(reticulate.conda_binary = "/opt/conda/bin/conda", SCP_env_name = "python-scp")
SCP::PrepareEnv(
miniconda_repo = "https://mirrors.bfsu.edu.cn/anaconda/miniconda",
pip_options = "-i https://pypi.tuna.tsinghua.edu.cn/simple"
)
准备数据
- 二代测序数据格式
-
wget https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz tar -zxvf pbmc3k_filtered_gene_bc_matrices.tar.gz
读入数据
pbmc <- Seurat::Read10X('~/data/filtered_gene_bc_matrices/hg19/')
sce <- Seurat::CreateSeuratObject(pbmc, project = 'pbmc3k',
min.cells = 3, min.features = 200)
# rm(pbmc);gc()
# 线粒体基因
sce[["percent.mt"]] <- Seurat::PercentageFeatureSet(sce, pattern = "^MT-")
# 核糖体基因
sce[["percent.ribo"]] <- Seurat::PercentageFeatureSet(sce, pattern = "^RP(S|L)")
# 血红蛋白基因 以 "HB" 开头,并且后面紧跟的字符不是 "P"
sce[["percent.hb"]] <- Seurat::PercentageFeatureSet(sce, pattern = "^HB[^(P)]")
sce@meta.data
环境 RNA 校正
decontX_results <- decontX::decontX(sce@assays$RNA@counts)
sce$Contamination = decontX_results$contamination
sce@meta.data
计算细胞周期评分
sce <- Seurat::NormalizeData(sce)
g2m_genes <- Seurat::CaseMatch(search=Seurat::cc.genes$g2m.genes,
match=rownames(sce))
s_genes <- Seurat::CaseMatch(search=Seurat::cc.genes$s.genes,
match=rownames(sce))
sce <- Seurat::CellCycleScoring(sce, g2m.features=g2m_genes, s.features=s_genes)
sce$CC.Difference <- sce$S.Score - sce$G2M.Score
双细胞过滤
-
sce <- Seurat::SCTransform(sce, vst.flavor = "v2", vars.to.regress = c("percent.mt", "percent.ribo", "percent.hb", "Contamination", "CC.Difference"), verbose = F)sweep.res <- DoubletFinder::paramSweep_v3(sce, PCs = 1:10, sct = T, num.cores=8) sweep.stats <- DoubletFinder::summarizeSweep(sweep.res, GT = FALSE) bcmvn <- DoubletFinder::find.pK(sweep.stats)## Assuming 1.6% doublet formation rate - tailor for your dataset nExp_poi <- round(0.016*nrow(sce@meta.data)) pK_bcmvn <- as.numeric(as.character(bcmvn$pK[which.max(bcmvn$BCmetric)])) sce <- DoubletFinder::doubletFinder_v3(sce, PCs = 1:10, pN = 0.25, pK = pK_bcmvn, nExp = nExp_poi, reuse.pANN = FALSE, sct = T) sce@meta.data
可视化
sce <- Seurat::RunPCA(sce, assay="SCT", verbose = FALSE)
Seurat::ElbowPlot(sce, ndims = 50)
sce <- Seurat::RunUMAP(sce, reduction = "pca",
dims = 1:30, verbose = FALSE)
sce <- Seurat::FindNeighbors(sce, reduction = "pca",
dims = 1:30, verbose = FALSE)
options(repr.plot.width = 8, repr.plot.height = 12)
FeaturePlot(sce,
features = c("percent.mt", "percent.ribo", "percent.hb", "Contamination", "CC.Difference"),
raster=FALSE # 细胞过多时候需要加这个参数
)
options(repr.plot.width = 6, repr.plot.height = 6)
DimPlot(sce, reduction = "umap", group.by = c("DF.classifications_0.25_0.05_43"))
options(repr.plot.width = 12, repr.plot.height = 6)
Seurat::VlnPlot(sce, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
plot1 <- Seurat::FeatureScatter(sce, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- Seurat::FeatureScatter(sce, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
过滤低质量细胞
sce <- subset(sce, DF.classifications_0.25_0.04_43 != 'Doublet'
& Contamination < 0.2
)
查看分群走向
sce <- Seurat::SCTransform(sce, vst.flavor = "v2",
vars.to.regress = c("percent.mt", "percent.ribo", "percent.hb", "Contamination", "CC.Difference"),
verbose = F)
sce <- Seurat::RunPCA(sce, assay="SCT", verbose = FALSE)
Seurat::ElbowPlot(sce, ndims = 50)
sce <- Seurat::RunUMAP(sce, reduction = "pca",
dims = 1:12, verbose = FALSE)
sce <- Seurat::FindNeighbors(sce, reduction = "pca",
dims = 1:12, verbose = FALSE)
sce <- Seurat::FindClusters(
object = sce,
resolution = c(seq(.1,1.6,.2)) #起始粒度,结束粒度,间隔
)
options(repr.plot.width = 12, repr.plot.height = 12)
require(clustree)
clustree::clustree(sce@meta.data, prefix = "SCT_snn_res.")
options(repr.plot.width = 6, repr.plot.height = 6)
DimPlot(sce, reduction = "umap", group.by = c("SCT_snn_res.0.9"))
可视化
sub_celltype <- c("0","1" ,"2" ,"3")
sub.celltype_list <- sapply(sub_celltype,function(slide){
print(slide)
sub.celltype <- subset(sce , `SCT_snn_res.0.1` == slide )
sub.celltype <- Seurat::SCTransform(sub.celltype, vst.flavor = "v2",
vars.to.regress = c("percent.mt", "percent.ribo", "percent.hb", "Contamination", "CC.Difference"),
verbose = F)
sub.celltype <- RunPCA(sub.celltype, npcs = 20)
sub.celltype <- FindNeighbors(sub.celltype, dims = 1:20)
sub.celltype <- FindClusters(sub.celltype, resolution = 0.9)
sub.celltype <- RunUMAP(sub.celltype, dims = 1:20)
return(sub.celltype)
})
sub.celltype_list
require(tidyverse)
require(plot1cell)
my36colors <-c('#E5D2DD', '#53A85F', '#F1BB72', '#F3B1A0', '#D6E7A3', '#57C3F3', '#476D87',
'#E95C59', '#E59CC4', '#AB3282', '#23452F', '#BD956A', '#8C549C', '#585658',
'#9FA3A8', '#E0D4CA', '#5F3D69', '#C5DEBA', '#58A4C3', '#E4C755', '#F7F398',
'#AA9A59', '#E63863', '#E39A35', '#C1E6F3', '#6778AE', '#91D0BE', '#B53E2B',
'#712820', '#DCC1DD', '#CCE0F5', '#CCC9E6', '#625D9E', '#68A180', '#3A6963',
'#968175'
)
plot_circlize_change <- function (data_plot, do.label = T, contour.levels = c(0.2, 0.3),
pt.size = 0.5, kde2d.n = 1000, contour.nlevels = 100, bg.color = "#F9F2E4",
col.use = NULL, label.cex = 0.5, labels.cex = 0.5, circos.cex = 0.5 ,repel = FALSE)
{
centers <- data_plot %>% dplyr::group_by(Cluster) %>% summarise(x = median(x = x),
y = median(x = y))
z <- MASS::kde2d(data_plot$x, data_plot$y, n = kde2d.n)
celltypes <- names(table(data_plot$Cluster))
cell_colors <- (scales::hue_pal())(length(celltypes))
if (!is.null(col.use)) {
cell_colors = col.use
col_df <- data.frame(Cluster = celltypes, color2 = col.use)
cells_order <- rownames(data_plot)
data_plot <- merge(data_plot, col_df, by = "Cluster")
rownames(data_plot) <- data_plot$cells
data_plot <- data_plot[cells_order, ]
data_plot$Colors <- data_plot$color2
}
circos.clear()
par(bg = bg.color)
circos.par(cell.padding = c(0, 0, 0, 0), track.margin = c(0.01,
0), track.height = 0.01, gap.degree = c(rep(2, (length(celltypes) -
1)), 12), points.overflow.warning = FALSE)
circos.initialize(sectors = data_plot$Cluster, x = data_plot$x_polar2)
circos.track(data_plot$Cluster, data_plot$x_polar2, y = data_plot$dim2,
bg.border = NA, panel.fun = function(x, y) {
circos.text(CELL_META$xcenter, CELL_META$cell.ylim[2] +
mm_y(4), CELL_META$sector.index, cex = labels.cex,
col = "black", facing = "bending.inside", niceFacing = T)
#circos.axis(labels.cex = 0.3, col = "black", labels.col = "black")
circos.axis(labels.cex = circos.cex, col = "black", labels.col = "black")
})
for (i in 1:length(celltypes)) {
dd <- data_plot[data_plot$Cluster == celltypes[i], ]
circos.segments(x0 = min(dd$x_polar2), y0 = 0, x1 = max(dd$x_polar2),
y1 = 0, col = cell_colors[i], lwd = 3, sector.index = celltypes[i])
}
text(x = 1, y = 0.1, labels = "Cluster", cex = 0.4, col = "black",
srt = -90)
points(data_plot$x, data_plot$y, pch = 19, col = alpha(data_plot$Colors,
0.9), cex = pt.size)
contour(z, drawlabels = F, nlevels = 100, levels = contour.levels,
col = "#ae9c76", add = TRUE)
if (do.label) {
if (repel) {
textplot(x = centers$x, y = centers$y, words = centers$Cluster,
cex = label.cex, new = F, show.lines = F)
}
else {
text(centers$x, centers$y, labels = centers$Cluster,
cex = label.cex, col = "black")
}
}
}
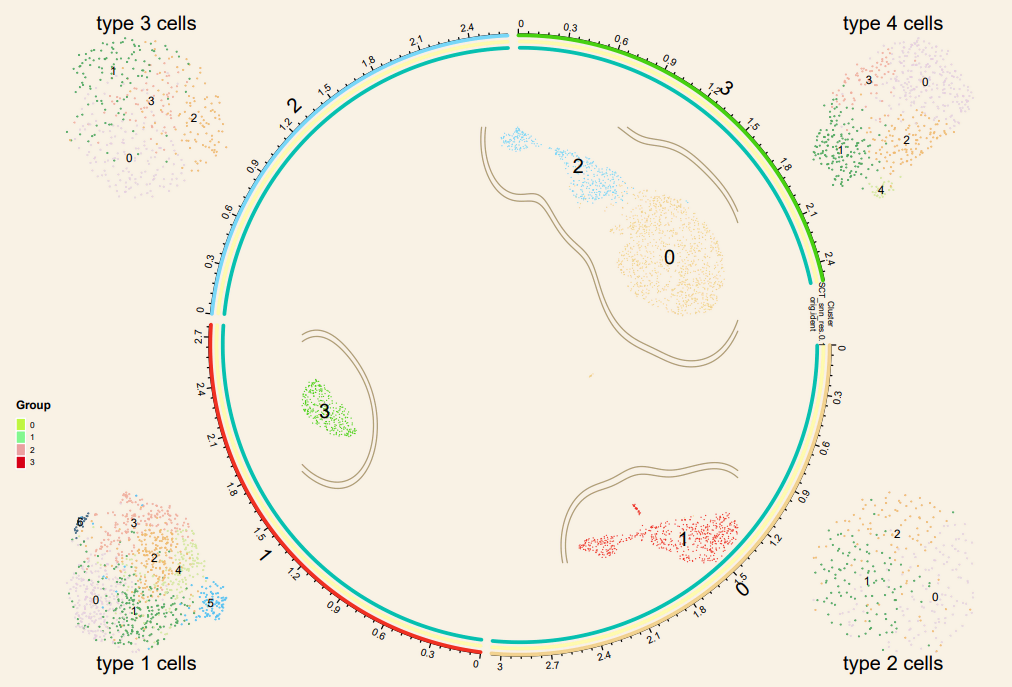
###Prepare data for ploting 准备圈图数据
circ_data <- plot1cell::prepare_circlize_data(sce, scale = 0.7) # 画出框了就调小 scale
set.seed(1234)
# 设置细胞分群信息的颜色
cluster_colors <- circlize::rand_color(length(levels(sce)))
group_colors <- circlize::rand_color(length(names(table(sce$`SCT_snn_res.0.1`))))
rep_colors <- circlize::rand_color(length(names(table(sce$orig.ident))))
# 绘制细胞分群圈图
{pdf(file = 'umap_circlize.plot.pdf', width = 9, height = 6) # 导出PDF开始
options(repr.plot.width = 12, repr.plot.height = 8)
plot_circlize_change(circ_data,do.label = T, pt.size = 0.01,
col.use = cluster_colors ,
bg.color = '#F9F2E5',
kde2d.n = 1000,
repel = T,
labels.cex = 1,
circos.cex = 0.5,
label.cex = 1)
plot1cell::add_track(circ_data,
group = "SCT_snn_res.0.1",
colors = group_colors, track_num = 2) ## can change it to one of the columns in the meta data of your seurat object
plot1cell::add_track(circ_data,
group = "orig.ident",
colors = rep_colors, track_num = 3)
# 左下
sub_index = 1
subcolors <- my36colors[1:nlevels(sub.celltype_list[[sub_index]])]
sub_1_meta<-get_metadata(sub.celltype_list[[sub_index]], color = subcolors)
sub_1_meta %>%
dplyr::group_by(seurat_clusters) %>%
summarize(x = median(x = x),y = median(x = y)) -> centers_1
points(sub_1_meta$x*0.32-1.2,sub_1_meta$y*0.32-0.73, pch = 19, col = alpha(sub_1_meta$Colors,0.9), cex = 0.1);
text(centers_1$x*0.32-1.2,centers_1$y*0.32-0.73, labels=centers_1$seurat_clusters, cex = 0.6, col = 'black')
# 右上
sub_index = 2
subcolors <- my36colors[1:nlevels(sub.celltype_list[[sub_index]])]
sub_1_meta<-get_metadata(sub.celltype_list[[sub_index]], color = subcolors)
sub_1_meta %>%
dplyr::group_by(seurat_clusters) %>%
summarize(x = median(x = x),y = median(x = y)) -> centers_1
points(sub_1_meta$x*0.32+1.2,sub_1_meta$y*0.32+0.73, pch = 19, col = alpha(sub_1_meta$Colors,0.9), cex = 0.1);
text(centers_1$x*0.32+1.2,centers_1$y*0.32+0.73, labels=centers_1$seurat_clusters, cex = 0.6, col = 'black')
# 左上
sub_index = 3
subcolors <- my36colors[1:nlevels(sub.celltype_list[[sub_index]])]
sub_1_meta<-get_metadata(sub.celltype_list[[sub_index]], color = subcolors)
sub_1_meta %>%
dplyr::group_by(seurat_clusters) %>%
summarize(x = median(x = x),y = median(x = y)) -> centers_1
points(sub_1_meta$x*0.32-1.2,sub_1_meta$y*0.32+0.73, pch = 19, col = alpha(sub_1_meta$Colors,0.9), cex = 0.1);
text(centers_1$x*0.32-1.2,centers_1$y*0.32+0.73, labels=centers_1$seurat_clusters, cex = 0.6, col = 'black')
# 右下
sub_index = 4
subcolors <- my36colors[1:nlevels(sub.celltype_list[[sub_index]])]
sub_1_meta<-get_metadata(sub.celltype_list[[sub_index]], color = subcolors)
sub_1_meta %>%
dplyr::group_by(seurat_clusters) %>%
summarize(x = median(x = x),y = median(x = y)) -> centers_1
points(sub_1_meta$x*0.32+1.2,sub_1_meta$y*0.32-0.73, pch = 19, col = alpha(sub_1_meta$Colors,0.9), cex = 0.1);
text(centers_1$x*0.32+1.2,centers_1$y*0.32-0.73, labels=centers_1$seurat_clusters, cex = 0.6, col = 'black')
title_text <- function(x0, y0, x1, y1, text, rectArgs = NULL, textArgs = NULL) {
center <- c(mean(c(x0, x1)), mean(c(y0, y1)))
do.call('rect', c(list(xleft = x0, ybottom = y0, xright = x1, ytop = y1), rectArgs))
do.call('text', c(list(x = center[1], y = center[2], labels = text), textArgs))
}
# 四周的title
title_text(x0 = -1.35, x1 = -1.05, y0 = -1.06, y1=-1, text = 'type 1 cells',
rectArgs = list(border='#F9F2E4',lwd=0.5),
textArgs = list(col='black',cex = 1))
title_text(x0 = 1.05, x1 = 1.35, y0 = -1.06, y1=-1, text = 'type 2 cells',
rectArgs = list(border='#F9F2E4',lwd=0.5),
textArgs = list(col='black',cex = 1))
title_text(x0 = -1.35, x1 = -1.05, y0 = 1.06, y1=1, text = 'type 3 cells',
rectArgs = list(border='#F9F2E4',lwd=0.5),
textArgs = list(col='black',cex = 1))
title_text(x0 = 1.05, x1 = 1.35, y0 = 1.06, y1=1, text = 'type 4 cells',
rectArgs = list(border='#F9F2E4',lwd=0.5),
textArgs = list(col='black',cex = 1))
#plot group#
Idents(sce) <- "SCT_snn_res.0.1"
col_use <- my36colors[1:nlevels(sce)]
cc <- get_metadata(sce, color = col_use)
cc %>%
dplyr::group_by("SCT_snn_res.0.1") %>%
summarize(x = median(x = x),y = median(x = y)) -> centers
col_group <- c('#bff542','#83f78f','#EBA1A2','#D70016')
lgd_points = Legend(labels = names(table(cc$`SCT_snn_res.0.1`)), type = "points",
title_position = "topleft",
title = "Group",
title_gp = gpar(col='black',fontsize = 7, fontface='bold'),
legend_gp = gpar(col = col_group),
labels_gp = gpar(col='black',fontsize = 5),
grid_height = unit(2, "mm"),
grid_width = unit(2, "mm"),
background = col_group)
draw(lgd_points, x = unit(15, "mm"), y = unit(50, "mm"),
just = c("right", "bottom"))
dev.off()} # 导出 PDF 结束