
2023-09-24-【学习】使用GATK4-0找SNP.md 23 KB
title: 【学习】使用GATK4.0找SNP urlname: shi-yong-GATK-zhao-SNP date: 2023-09-24 18:49:18 index_img: https://api.limour.top/randomImg?d=2023-09-24 18:49:18
tags: ['GATK', 'SNP', 'WGS']
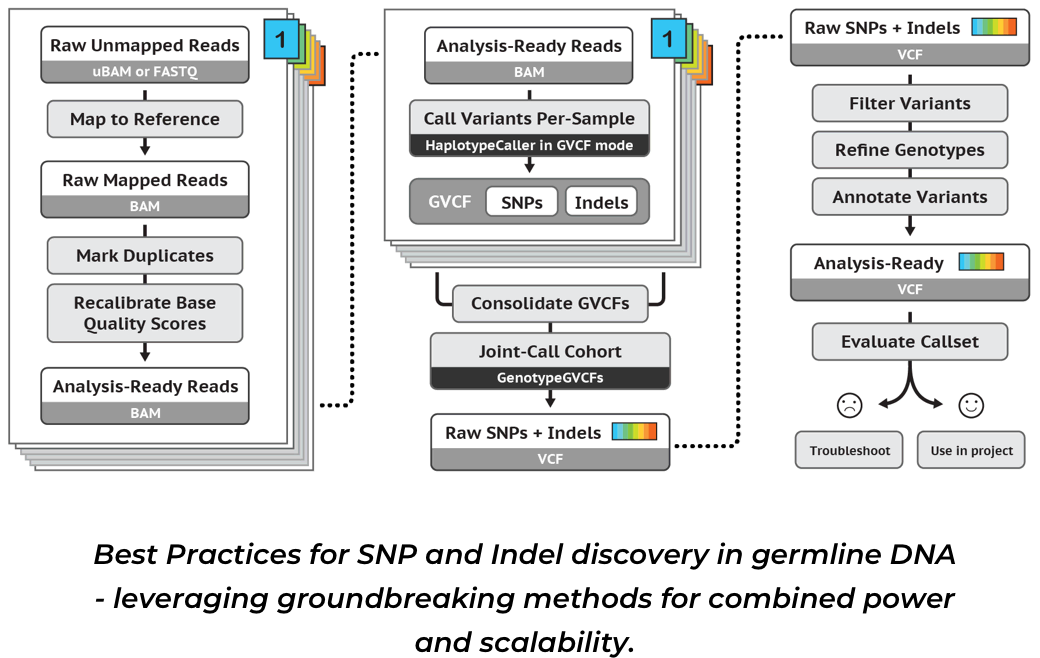
配置环境
- 基础编程环境
- GitHub 下载加速
-
SRA工具
conda create -n sra_tools -c bioconda sra-tools conda activate sra_tools conda install -c conda-forge lftp -y conda install -c conda-forge pigz -y # 或许换成 pbgzip 更好,此时将 -p 换成 -n 来指定线程数 conda install -c bioconda pbgzip -y prefetch # vdb-config -i # 设置 HTTP 代理GATK4
conda create -n GATK4 -c bioconda gatk4 conda activate GATK4 conda install -c bioconda samtools -y conda install -c bioconda bwa -y conda install -c bioconda pbgzip -y # 并行版bgzip,bgzip是修改过的gzip,更适合生信领域 conda install -c bioconda tabix -y # 操作VCF文件,与bgzip配套 # conda install -c bioconda fastqc -y # 改用fastp了 # conda install -c bioconda trimmomatic -y # 改用fastp了 conda install -c bioconda fastp -y # conda install -c bioconda bcftools -y # 用于重命名染色体 # ln -s $CONDA_PREFIX/lib/libgsl.so $CONDA_PREFIX/lib/libgsl.so.25 # 无效,放弃 # conda create -n GATK4-VEP -c bioconda ensembl-vep -y # 根正苗红的突变注释软件,不懂有什么奇怪依赖,解析环境半天 BWA是DNA比对工具(不会跨外显子比对),STAR是RNA比对工具
-
准备数据
参考数据
NCBI上各物种的参考序列,可以找到RefSeq,比如Human是GCF_000001405
知道序号后可以到FTP上下载相应的
genomic.fna.gz文件比如GCF_000001405,依次进入
000/001/405即可找到对应的文件wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.40_GRCh38.p14/GCF_000001405.40_GRCh38.p14_genomic.fna.gz -O GRCh38.p14.fna.gz conda run -n sra_tools pigz -d GRCh38.p14.fna.gz # 得到 GRCh38.p14.fna # 创建索引 samtools faidx GRCh38.p14.fna # 得到 GRCh38.p14.fna.fai # 查看一段序列 samtools faidx GRCh38.p14.fna NC_000001.11:1000000-1000200 # 创建比对索引 bwa index GRCh38.p14.fna # 会自动在 bwtsw, is or rb2 三种算法中选择合适的 # 创建dict gatk CreateSequenceDictionary -R GRCh38.p14.fna最后得到的RefSeq目录结构如下
# conda create -n linux -c conda-forge tree # conda run -n linux tree -f -h --du [8.5G] . ├── [ 79K] ./GRCh38.p14.dict ├── [3.1G] ./GRCh38.p14.fna ├── [ 21K] ./GRCh38.p14.fna.amb ├── [ 90K] ./GRCh38.p14.fna.ann ├── [3.1G] ./GRCh38.p14.fna.bwt ├── [ 26K] ./GRCh38.p14.fna.fai ├── [786M] ./GRCh38.p14.fna.pac └── [1.5G] ./GRCh38.p14.fna.sa已知SNP
lftp ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/,密码空,直接回车下载和参考数据相对应的indels.hg38.vcf
wget https://ftp.ncbi.nlm.nih.gov/snp/latest_release/VCF/GCF_000001405.40.gz -O GRCh38.dbSNP.ncbi.vcf.gz转换染色体名称到NCBI的参考文件
assembly_report.txt在下载NCBI参考数据FTP目录下wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.40_GRCh38.p14/GCF_000001405.40_GRCh38.p14_assembly_report.txt -O GRCh38.p14_assembly_report.txt grep -e '^[^#]' GRCh38.p14_assembly_report.txt | awk -F'\t' '{ print $NF, $7 }' | sed 's/\r / /g' > rename_file.txt conda create -n something_fuck -c conda-forge mamba conda activate something_fuck mamba install -c bioconda bcftools bcftools annotate --rename-chrs rename_file.txt -o Homo_sapiens_assembly38.known_indels.ncbi.vcf Homo_sapiens_assembly38.known_indels.vcf bcftools annotate --rename-chrs rename_file.txt -o hapmap_3.3.hg38.ncbi.vcf hapmap_3.3.hg38.vcf bcftools annotate --rename-chrs rename_file.txt -o Mills_and_1000G_gold_standard.indels.hg38.ncbi.vcf Mills_and_1000G_gold_standard.indels.hg38.vcf conda run -n GATK4 pbgzip -n 4 *.ncbi.vcf # 似乎一次只压缩一个,多运行几次建立索引
nano knownSitesIndex.sh && chmod +x knownSitesIndex.sh ./knownSitesIndex.sh#!/bin/bash source activate GATK4 #设置knownSites数据存放目录 knownSites=/home/jovyan/upload/knownSites for file in $knownSites/*.ncbi.vcf.gz do echo $file gatk IndexFeatureFile \ -I $file done最后得到的knownSites目录结构如下
[ 25G] . ├── [ 25G] ./GRCh38.dbSNP.ncbi.vcf.gz ├── [4.2M] ./GRCh38.dbSNP.ncbi.vcf.gz.tbi ├── [ 79K] ./GRCh38.p14_assembly_report.txt ├── [ 61M] ./hapmap_3.3.hg38.ncbi.vcf.gz ├── [2.1M] ./hapmap_3.3.hg38.ncbi.vcf.gz.tbi ├── [ 58M] ./Homo_sapiens_assembly38.known_indels.ncbi.vcf.gz ├── [2.1M] ./Homo_sapiens_assembly38.known_indels.ncbi.vcf.gz.tbi ├── [ 20M] ./Mills_and_1000G_gold_standard.indels.hg38.ncbi.vcf.gz ├── [2.0M] ./Mills_and_1000G_gold_standard.indels.hg38.ncbi.vcf.gz.tbi └── [ 23K] ./rename_file.txt测序数据
WGS的DNA测序数据
下载方式见SRA文件转FASTQ文件
也可以到ENA数据库上下载
conda run -n sra_tools prefetch --option-file SRR_Acc_List.txt nano 11.sh && chmod +x 11.sh ./11.sh#!/bin/bash source activate sra_tools #任务名 TASKN=SRX247249 #设置SRA根目录, pwd是当前目录 ROOTDIR=`pwd` #设置rawData存放目录 rawData=/home/jovyan/upload/rawData/$TASKN mkdir -p $rawData cd $ROOTDIR for file in `cat SRR_Acc_List.txt` do echo $file mkdir $rawData/$file cd $rawData/$file fasterq-dump --split-3 $ROOTDIR/$file -e 6 pigz -p 6 * done
rawData质控
- 原始数据质量判断
-
质量判断(可跳过)
nano qc.sh && chmod +x qc.sh ./qc.sh#!/bin/bash source activate GATK4 #任务名 TASKN=SRX247249 #设置rawData存放目录 rawData=/home/jovyan/upload/rawData/$TASKN #设置qc结果的输出目录 QCDIR=/home/jovyan/upload/rawData/$TASKN"_fastqc" mkdir -p $QCDIR for file in $rawData/* do echo $file SAMPLE=${file##*/} echo $QCDIR"/"$SAMPLE mkdir $QCDIR"/"$SAMPLE fastqc -o $QCDIR"/"$SAMPLE --threads=6 `ls $rawData/$SAMPLE/*` done 对于PE而言,正向和反向reads的测量过程是独立的,将当成两次SE来处理
最后的报告中:
Q20的碱基要在95%以上(最差不低于90%)
Q30要求大于85%(最差也不要低于80%)
对于人类来说,GC含量应该在40%左右
fastp一键质控
-
nano qc.sh && chmod +x qc.sh ./qc.sh#!/bin/bash source activate GATK4 #任务名 TASKN=SRX247249 #设置rawData存放目录 rawData=/home/jovyan/upload/rawData/$TASKN #设置qc结果的输出目录 QCDIR=/home/jovyan/upload/rawData/$TASKN"_fastp" mkdir -p $QCDIR #设置cleanData的存放目录 CLEAN=/home/jovyan/upload/cleanData/$TASKN mkdir -p $CLEAN for file in $rawData/* do echo $file SAMPLE=${file##*/} echo $QCDIR"/"$SAMPLE mkdir $QCDIR"/"$SAMPLE echo $CLEAN"/"$SAMPLE mkdir $CLEAN"/"$SAMPLE cd $QCDIR"/"$SAMPLE fastp -c -w 4 \ -o $CLEAN"/"$SAMPLE"/out.R1.fq.gz" \ -O $CLEAN"/"$SAMPLE"/out.R2.fq.gz" \ -h $QCDIR"/"$SAMPLE"/fastp.html" \ -j $QCDIR"/"$SAMPLE"/fastp.json" \ -i `ls $rawData/$SAMPLE/*_1.fastq.gz` \ -I `ls $rawData/$SAMPLE/*_2.fastq.gz` done 最后得到的cleanData目录结构如下
[ 23G] . ├── [9.1G] ./SRR799559 │ ├── [4.4G] ./SRR799559/out.R1.fq.gz │ └── [4.7G] ./SRR799559/out.R2.fq.gz ├── [7.0G] ./SRR799560 │ ├── [3.4G] ./SRR799560/out.R1.fq.gz │ └── [3.5G] ./SRR799560/out.R2.fq.gz └── [7.4G] ./SRR799561 ├── [3.6G] ./SRR799561/out.R1.fq.gz └── [3.8G] ./SRR799561/out.R2.fq.gz数据比对
nano bwa_and_markdup.sh && chmod +x bwa_and_markdup.sh ./bwa_and_markdup.sh#!/bin/bash source activate GATK4 #任务名 TASKN=SRX247249 #设置cleanData的存放目录 CLEAN=/home/jovyan/upload/cleanData/$TASKN #设置RefSeq的存放目录 RefSeq=/home/jovyan/data/refseq/GRCh38.p14.fna #设置Read Group信息,见 https://gatk.broadinstitute.org/hc/en-us/articles/360035890671-Read-groups RGroup_PL=ILLUMINA # 所用的测序平台:ILLUMINA,SLX,SOLEXA,SOLID,454,LS454,COMPLETE,PACBIO,IONTORRENT,CAPILLARY,HELICOS或UNKNOWN。CG测序为COMPLETE RGroup_SM=$TASKN # 样本ID,同一个样本可能有多个lane,此时用样本ID相关联 RGroup='PL:'$RGroup_PL'\tSM:'$RGroup_SM #设置BAM的存放目录 BAM=/home/jovyan/upload/BAM/$TASKN mkdir -p $BAM for file in $CLEAN/* do echo $file SAMPLE=${file##*/} echo $BAM"/"$SAMPLE mkdir $BAM"/"$SAMPLE echo '@RG\tID:'$SAMPLE'\t'$RGroup #1 比对 bwa mem -t 4 -M -R '@RG\tID:'$SAMPLE'\t'$RGroup $RefSeq `ls $CLEAN/$SAMPLE/*` \ | samtools view -Sb - > $BAM"/"$SAMPLE"/raw.bam" #2 排序 samtools sort -@ 4 -m 4G -O bam -o $BAM"/"$SAMPLE"/sorted.bam" $BAM"/"$SAMPLE"/raw.bam" rm $BAM"/"$SAMPLE"/raw.bam" #3 标记PCR重复 gatk MarkDuplicates -I $BAM"/"$SAMPLE"/sorted.bam" \ -O $BAM"/"$SAMPLE"/sorted.markdup.bam" \ -M $BAM"/"$SAMPLE"/sorted.markdup_metrics.txt" rm $BAM"/"$SAMPLE"/sorted.bam" #4 创建比对索引文件 samtools index $BAM"/"$SAMPLE"/sorted.markdup.bam" done最后得到的BAM目录结构如下
[ 30G] . ├── [ 11G] ./SRR799559 │ ├── [ 11G] ./SRR799559/sorted.markdup.bam │ ├── [4.5M] ./SRR799559/sorted.markdup.bam.bai │ └── [3.7K] ./SRR799559/sorted.markdup_metrics.txt ├── [8.8G] ./SRR799560 │ ├── [8.8G] ./SRR799560/sorted.markdup.bam │ ├── [3.9M] ./SRR799560/sorted.markdup.bam.bai │ └── [3.7K] ./SRR799560/sorted.markdup_metrics.txt └── [9.6G] ./SRR799561 ├── [9.5G] ./SRR799561/sorted.markdup.bam ├── [4.1M] ./SRR799561/sorted.markdup.bam.bai └── [3.7K] ./SRR799561/sorted.markdup_metrics.txt同样本合并
nano merge.sh && chmod +x merge.sh ./merge.sh#!/bin/bash source activate GATK4 #任务名 TASKN=SRX247249 #设置BAM的存放目录 BAM=/home/jovyan/upload/BAM/$TASKN #设置merge后的数据存放目录 MERGEDBAM=/home/jovyan/upload/merged/$TASKN/SAMPLE1 mkdir -p $MERGEDBAM samtools merge $MERGEDBAM"/sorted.markdup.bam" \ `find "$BAM" -name "sorted.markdup.bam" -type f -exec readlink -f {} \;` samtools index $MERGEDBAM"/sorted.markdup.bam"最后得到的MERGED目录结构如下
[ 21G] . └── [ 21G] ./SAMPLE1 ├── [ 21G] ./SAMPLE1/sorted.markdup.bam └── [6.8M] ./SAMPLE1/sorted.markdup.bam.bai局部重比对
具体见黄树嘉博士的相关介绍
因为本文是GATK 4.0的HaplotypeCaller模块,自带局部重比对,故用到的时候再写
BQSR
nano BQSR.sh && chmod +x BQSR.sh ./BQSR.sh#!/bin/bash source activate GATK4 #任务名 TASKN=SRX247249 #设置merged数据存放目录 MERGED=/home/jovyan/upload/merged/$TASKN #设置RefSeq的存放目录 RefSeq=/home/jovyan/data/refseq/GRCh38.p14.fna #设置knownSites数据存放目录 knownSites=/home/jovyan/upload/knownSites knownSites=$(echo $(ls $knownSites/*.ncbi.vcf.gz | sed 's/^/--known-sites /' | tr '\n' ' ')) echo $knownSites for file in $MERGED/* do echo $file SAMPLE=${file##*/} echo $MERGED"/"$SAMPLE gatk BaseRecalibrator $knownSites \ -R $RefSeq \ -I $MERGED"/"$SAMPLE"/sorted.markdup.bam" \ -O $MERGED"/"$SAMPLE"/recal_data.table" gatk ApplyBQSR \ -R $RefSeq \ -I $MERGED"/"$SAMPLE"/sorted.markdup.bam" \ --bqsr-recal-file $MERGED"/"$SAMPLE"/recal_data.table" \ -O $MERGED"/"$SAMPLE"/sorted.markdup.BQSR.bam" samtools index $MERGED"/"$SAMPLE"/sorted.markdup.BQSR.bam" done最后得到的MERGED目录结构如下
[ 54G] . └── [ 54G] ./SAMPLE1 ├── [2.5M] ./SAMPLE1/recal_data.table ├── [ 21G] ./SAMPLE1/sorted.markdup.bam ├── [6.8M] ./SAMPLE1/sorted.markdup.bam.bai ├── [8.8M] ./SAMPLE1/sorted.markdup.BQSR.bai ├── [ 33G] ./SAMPLE1/sorted.markdup.BQSR.bam └── [7.6M] ./SAMPLE1/sorted.markdup.BQSR.bam.bai两步法变异检测
HaplotypeCaller
nano HaplotypeCaller.sh && chmod +x HaplotypeCaller.sh ./HaplotypeCaller.sh#!/bin/bash source activate GATK4 #任务名 TASKN=SRX247249 #设置BQSR数据存放目录 MERGED=/home/jovyan/upload/merged/$TASKN #设置RefSeq的存放目录 RefSeq=/home/jovyan/data/refseq/GRCh38.p14.fna for file in $MERGED/* do echo $file SAMPLE=${file##*/} echo $MERGED"/"$SAMPLE gatk --java-options "-Xmx4g" HaplotypeCaller -ERC GVCF \ -R $RefSeq \ -I $MERGED"/"$SAMPLE"/sorted.markdup.BQSR.bam" \ -O $MERGED"/"$SAMPLE"/HC.g.vcf.gz" done
CombineGVCFs
单样本
VCFPATH=$MERGED'/SAMPLE1'
多样本
nano CombineGVCFs.sh && chmod +x CombineGVCFs.sh
./CombineGVCFs.sh
#!/bin/bash
source activate GATK4
#任务名
TASKN=SRX247249
#设置BQSR数据存放目录
MERGED=/home/jovyan/upload/merged/$TASKN
#设置RefSeq的存放目录
RefSeq=/home/jovyan/data/refseq/GRCh38.p14.fna
#设置最后输出的路径
VCFPATH=/home/jovyan/upload/VCF/$TASKN
mkdir -p $VCFPATH
variant=$(echo $(ls $MERGED/*/HC.g.vcf.gz | sed 's/^/--variant /' | tr '\n' ' '))
echo $variant
gatk CombineGVCFs $$variant \
-R $RefSeq \
-O $VCFPATH'/HC.g.vcf.gz'
GenotypeGVCFs
nano GenotypeGVCFs.sh && chmod +x GenotypeGVCFs.sh
./GenotypeGVCFs.sh
#!/bin/bash
source activate GATK4
#任务名
TASKN=SRX247249
#设置BQSR数据存放目录
MERGED=/home/jovyan/upload/merged/$TASKN
#设置RefSeq的存放目录
RefSeq=/home/jovyan/data/refseq/GRCh38.p14.fna
#设置最后输出的路径
VCFPATH=$MERGED'/SAMPLE1'
mkdir -p $VCFPATH
gatk --java-options "-Xmx4g" GenotypeGVCFs \
-R $RefSeq \
-V $VCFPATH'/HC.g.vcf.gz' \
-O $VCFPATH'/HC.vcf.gz'
最后得到的结果如下
├── [6.8G] ./HC.g.vcf.gz ├── [5.0M] ./HC.g.vcf.gz.tbi ├── [127M] ./HC.vcf.gz ├── [2.0M] ./HC.vcf.gz.tbiVQSR
nano VQSR.sh && chmod +x VQSR.sh ./VQSR.sh#!/bin/bash source activate GATK4 #任务名 TASKN=SRX247249 #设置BQSR数据存放目录 MERGED=/home/jovyan/upload/merged/$TASKN #设置RefSeq的存放目录 RefSeq=/home/jovyan/data/refseq/GRCh38.p14.fna #设置最后输出的路径 VCFPATH=$MERGED'/SAMPLE1' #设置knownSites数据存放目录 knownSites=/home/jovyan/upload/knownSites gatk VariantRecalibrator \ -R $RefSeq \ -V $VCFPATH'/HC.vcf.gz' \ --resource:hapmap,known=false,training=true,truth=true,prior=15.0 $knownSites/hapmap_3.3.hg38.ncbi.vcf.gz \ --resource:dbsnp,known=true,training=false,truth=false,prior=2.0 $knownSites/GRCh38.dbSNP.ncbi.vcf.gz \ -an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR \ -mode SNP \ -O $VCFPATH/snp.recal \ --tranches-file $VCFPATH/snp.tranches \ --rscript-file $VCFPATH/snp.plots.R gatk ApplyVQSR \ -R $RefSeq \ -V $VCFPATH'/HC.vcf.gz' \ -O $VCFPATH'/snp.VQSR.vcf.gz' \ --truth-sensitivity-filter-level 99.0 \ --tranches-file $VCFPATH/snp.tranches \ --recal-file $VCFPATH/snp.recal \ -mode SNP gatk VariantRecalibrator \ -R $RefSeq \ -V $VCFPATH'/snp.VQSR.vcf.gz' \ --resource:dbindel,known=true,training=false,truth=false,prior=2.0 $knownSites/Homo_sapiens_assembly38.known_indels.ncbi.vcf.gz \ --resource:mills,known=true,training=true,truth=true,prior=12.0 $knownSites/Mills_and_1000G_gold_standard.indels.hg38.ncbi.vcf.gz \ -an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR \ -mode INDEL --max-gaussians 6 \ -O $VCFPATH/snp.indel.recal \ --tranches-file $VCFPATH/snp.indel.tranches \ --rscript-file $VCFPATH/snp.indel.plots.R gatk ApplyVQSR \ -R $RefSeq \ -V $VCFPATH'/snp.VQSR.vcf.gz' \ -O $VCFPATH'/snp.indel.VQSR.vcf.gz' \ --truth-sensitivity-filter-level 99.0 \ --tranches-file $VCFPATH/snp.indel.tranches \ --recal-file $VCFPATH/snp.indel.recal \ -mode INDEL最后得到的结果如下
├── [2.7M] ./snp.plots.R ├── [6.2M] ./snp.plots.R.pdf ├── [199M] ./snp.recal ├── [7.5M] ./snp.recal.idx ├── [ 584] ./snp.tranches ├── [7.5K] ./snp.tranches.pdf ├── [151M] ./snp.VQSR.vcf.gz ├── [2.0M] ./snp.VQSR.vcf.gz.tbi ├── [2.8M] ./snp.indel.plots.R ├── [6.2M] ./snp.indel.plots.R.pdf ├── [ 35M] ./snp.indel.recal ├── [256K] ./snp.indel.recal.idx ├── [ 595] ./snp.indel.tranches ├── [153M] ./snp.indel.VQSR.vcf.gz ├── [2.0M] ./snp.indel.VQSR.vcf.gz.tbiSNP内容示例
# tabix snp.VQSR.vcf.gz NC_000001.11 | head -n 5 NC_000001.11 16378 . T C 35.32 VQSRTrancheSNP99.90to100.00 AC=2;AF=1.00;AN=2;DP=2;ExcessHet=0.0000;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=20.00;QD=17.66;SOR=2.303;VQSLOD=-1.018e+01;culprit=MQ GT:AD:DP:GQ:PL 1/1:0,2:2:6:47,6,0 NC_000001.11 17020 . G A 59.32 VQSRTrancheSNP99.90to100.00 AC=2;AF=1.00;AN=2;DP=2;ExcessHet=0.0000;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=31.66;QD=29.66;SOR=0.693;VQSLOD=-5.480e+00;culprit=MQ GT:AD:DP:GQ:PL 1/1:0,2:2:6:71,6,0 NC_000001.11 17385 . G A 60.32 VQSRTrancheSNP99.90to100.00 AC=2;AF=1.00;AN=2;DP=2;ExcessHet=0.0000;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=32.28;QD=30.16;SOR=2.303;VQSLOD=-2.357e+00;culprit=MQ GT:AD:DP:GQ:PL 1/1:0,2:2:6:72,6,0 NC_000001.11 20254 . G A 64.64 VQSRTrancheSNP99.90to100.00 AC=1;AF=0.500;AN=2;BaseQRankSum=2.37;DP=8;ExcessHet=0.0000;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=24.89;MQRankSum=-2.030e-01;QD=8.08;ReadPosRankSum=-1.611e+00;SOR=1.034;VQSLOD=-1.317e+01;culprit=MQ GT:AD:DP:GQ:PGT:PID:PL:PS 0|1:5,3:8:72:1|0:20250_T_C:72,0,126:20250 NC_000001.11 39230 . G A 83.64 VQSRTrancheSNP99.90to100.00 AC=1;AF=0.500;AN=2;BaseQRankSum=-1.078e+00;DP=15;ExcessHet=0.0000;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=26.72;MQRankSum=2.20;QD=5.58;ReadPosRankSum=-1.917e+00;SOR=1.022;VQSLOD=-1.362e+01;culprit=MQ GT:AD:DP:GQ:PL 0/1:10,5:15:91:91,0,239变异注释
安装 VEP
-
sudo docker pull ensemblorg/ensembl-vep sudo docker run --rm -t -i -v ~/upload:/data:z ensemblorg/ensembl-vep pwd sudo docker run --rm -t -i -v ~/upload:/data:z ensemblorg/ensembl-vep ls -al /opt/vep/ sudo mkdir -p ~/upload/vep && sudo chmod 777 ~/upload/vep sudo docker run --rm -t -i -v ~/upload:/data:z -v ~/upload/vep:/opt/vep/.vep:z ensemblorg/ensembl-vep INSTALL.pl # sudo docker run --rm -t -i -v ~/upload:/data:Z ensemblorg/ensembl-vep cat INSTALL.pl > INSTALL.pl # 自行分析 INSTALL.pl,构造下载后的结构,以下是104版本的 # 太慢了,手动下载,请各显神通,下载地址来自上一步的输出 wget https://ftp.ensembl.org/pub/release-104/variation/indexed_vep_cache/homo_sapiens_vep_104_GRCh38.tar.gz -O ~/upload/vep tar -zxvf homo_sapiens_vep_104_GRCh38.tar.gz进行注释
mkdir -p ~/upload/VEP/SRX247249 && chmod -R 777 ~/upload/VEP/SRX247249 # mv ~/data/refseq ~/upload # chmod -R 777 ~/upload/refseq # chmod -R 777 ~/upload/vep # chmod -R 777 ~/upload/merged/SRX247249 sudo docker run --rm -t -i -v ~/upload:/data:z ensemblorg/ensembl-vep \ vep --fasta /data/refseq/GRCh38.p14.fna \ --format vcf --vcf --fork 4 --hgvs --force_overwrite --everything \ --offline --dir_cache /data/vep \ -i /data/merged/SRX247249/SAMPLE1/snp.indel.VQSR.vcf.gz \ -o /data/merged/SRX247249/SAMPLE1/snp.indel.VQSR.VEP.vcf # sudo chmod 777 ~/upload/merged/SRX247249/SAMPLE1/snp.indel.VQSR.VEP.vcf # pbgzip -n 4 snp.indel.VQSR.VEP.vcf # tabix -p vcf snp.indel.VQSR.VEP.vcf.gz 最后得到的结果如下
├── [346M] ./snp.indel.VQSR.VEP.vcf.gz ├── [210K] ./snp.indel.VQSR.VEP.vcf.gz_summary.html ├── [1.6M] ./snp.indel.VQSR.VEP.vcf.gz.tbi ├── [8.2K] ./snp.indel.VQSR.VEP.vcf.gz_warnings.txt# tabix snp.indel.VQSR.VEP.vcf.gz NC_000001.11 | head -n 2 NC_000001.11 16378 . T C 35.32 VQSRTrancheSNP99.90to100.00 AC=2;AF=1.00;AN=2;DP=2;ExcessHet=0.0000;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=20.00;QD=17.66;SOR=2.303;VQSLOD=-1.018e+01;culprit=MQ;CSQ=C|downstream_gene_variant|MODIFIER|DDX11L1|ENSG00000223972|Transcript|ENST00000450305|transcribed_unprocessed_pseudogene||||||||||rs148220436|2708|1||SNV|HGNC|HGNC:37102|YES||||||||||||||||||||||||||||||||||||||||||||,C|downstream_gene_variant|MODIFIER|DDX11L1|ENSG00000223972|Transcript|ENST00000456328|processed_transcript||||||||||rs148220436|1969|1||SNV|HGNC|HGNC:37102||||1|||||||||||||||||||||||||||||||||||||||||,C|intron_variant&non_coding_transcript_variant|MODIFIER|WASH7P|ENSG00000227232|Transcript|ENST00000488147|unprocessed_pseudogene||8/10|ENST00000488147.1:n.1067+229A>G|||||||rs148220436||-1||SNV|HGNC|HGNC:38034|YES||||||||||||||||||||||||||||||||||||||||||||,C|downstream_gene_variant|MODIFIER|MIR6859-1|ENSG00000278267|Transcript|ENST00000619216|miRNA||||||||||rs148220436|991|-1||SNV|HGNC|HGNC:50039|YES||||||||||||||||||||||||||||||||||||||||||||,C|regulatory_region_variant|MODIFIER|||RegulatoryFeature|ENSR00000344266|CTCF_binding_site||||||||||rs148220436||||SNV|||||||||||||||||||||||||||||||||||||||||||||||,C|regulatory_region_variant|MODIFIER|||RegulatoryFeature|ENSR00001164745|promoter_flanking_region||||||||||rs148220436||||SNV||||||||||||||||||||||||||||||||||||||||||||||| GT:AD:DP:GQ:PL 1/1:0,2:2:6:47,6,0 NC_000001.11 17020 . G A 59.32 VQSRTrancheSNP99.90to100.00 AC=2;AF=1.00;AN=2;DP=2;ExcessHet=0.0000;FS=0.000;MLEAC=1;MLEAF=0.500;MQ=31.66;QD=29.66;SOR=0.693;VQSLOD=-5.480e+00;culprit=MQ;CSQ=A|downstream_gene_variant|MODIFIER|DDX11L1|ENSG00000223972|Transcript|ENST00000450305|transcribed_unprocessed_pseudogene||||||||||rs199740902|3350|1||SNV|HGNC|HGNC:37102|YES||||||||||||||||||||||||||||||||||||||||||||,A|downstream_gene_variant|MODIFIER|DDX11L1|ENSG00000223972|Transcript|ENST00000456328|processed_transcript||||||||||rs199740902|2611|1||SNV|HGNC|HGNC:37102||||1|||||||||||||||||||||||||||||||||||||||||,A|non_coding_transcript_exon_variant|MODIFIER|WASH7P|ENSG00000227232|Transcript|ENST00000488147|unprocessed_pseudogene|7/11||ENST00000488147.1:n.746C>T||746|||||rs199740902||-1||SNV|HGNC|HGNC:38034|YES||||||||||||||||||||||||||||||||||||||||||||,A|downstream_gene_variant|MODIFIER|MIR6859-1|ENSG00000278267|Transcript|ENST00000619216|miRNA||||||||||rs199740902|349|-1||SNV|HGNC|HGNC:50039|YES|||||||||||||||||||||||||||||||||||||||||||| GT:AD:DP:GQ:PL 1/1:0,2:2:6:71,6,0